
Isidro Cortes-Ciriano@isidrolauscher
Nov 24, 2022
21 tweets
We are most happy to present SComatic, an algorithm for de novo detection of somatic mutations in high-throughput single-cell data, including single-cell RNAseq and ATAC-seq. Monumental effort led by @fmuyas biorxiv.org/content/10.110 Thread 
Context: detecting somatic mutations in single cells is critical to study somatic evolution, cell plasticity, cancer and clonal mosaicism. However, identifying mutations at single-cell resolution is challenging due to various types of artefacts (allelic imbalances, etc).
Detecting mutations in single cells and mapping them to cell phenotypes is even harder, as methods to perform joint profiling of transcriptomes and genotypes do not scale easily and require complex protocols.
Calling mutations in single-cell data sets directly is one appealing option (that is, calling mutations in scRNAseq reads). However, current methods (see below) cannot deal with the high error rates of single-cell data, which vastly limits mutation detection performance (below).
Thus, previous methods designed to detect somatic mutations in single-cell data rely on targeted amplification of well-studied cancer driver mutations (e.g. JAK2 V617F), or direct detection in sequencing reads of mutations identified using matched sequencing data (e.g. bulk WGS).
The key issue is that these approaches cannot be applied to existing data sets without matched bulk or single-cell DNAseq data, and are limited to study known mutations. As a result, the patterns and rates of mutations in most cell types remain largely unknown.
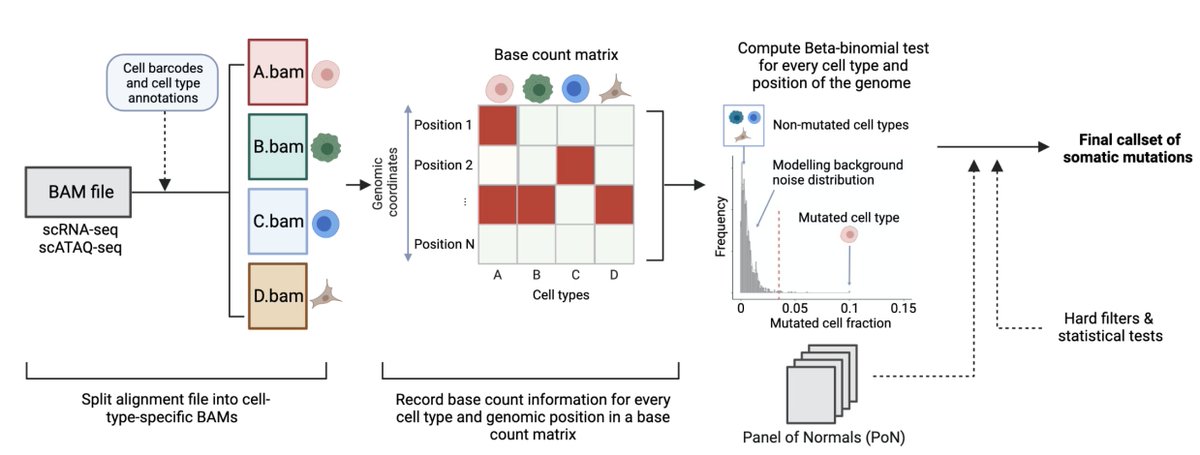
To address these limitations, we developed SComatic, an algorithm that permits detection of somatic mutations in single-cell data sets, including scRNAseq and scATACseq, without matched bulk or single-cell DNAseq data. Does it really work? How? Come and see
SComatic uses statistical models, filters, cell annotations and a "panel of normals" strategy to distinguish somatic mutations from technical and sequencing artefacts. Full details in the preprint and code here: github.com/cortes-ciriano

For validation purposes, we first used matched WES and scRNAseq data from cutaneous squamous cell carcinomas (pubmed.ncbi.nlm.nih.gov/32579974/). The mutations detected by SComatic in epithelial cells showed the spectrum expected for these samples (UV radiation):

Using these data, we show that the precision of SComatic is 20-40 times higher(!) than existing methods without compromising sensitivity! (this is one of the key plots in our study!!  )
)
) 
The higher performance of our approach is also reflected in the mutational spectra of the mutation call sets, with SComatic and WES detecting mutational signatures consistent with UV-light mutagenesis, which is in stark contrast to other methods:

This is even clearer if we have a look at the mutational spectra (see the cosine similarity with the WES data.. 0.99 for SComatic!):

We then called somatic mutations in epithelial cells from colorectal cancers (@NCI Human Tumor Atlas Network), which revealed mutational rates and signatures consistent with the underlying genotypes (e.g. high mutational burdens in MSI samples, which were comparable to TCGA data - see the preprint):

Mutational signature analysis allowed us to identify one POLE-deficient sample misclassified as MSI, which showed a clear contribution of mutational signatures associated with POLE-deficiency but not MMRD:

We also applied SComatic to scRNAseq data from #GTEx and sciATAC-seq data from the Ren's lab (pubmed.ncbi.nlm.nih.gov/34774128/), which allowed us to detect mutational signatures and rates of mutations at the single-cell and cell-type level across multiple organs.

In addition, we characterised the mutational rates and signatures in cardiomyocytes using data from the human cell atlas (@Teichlab), which were broadly consistent with recent single-cell WGS data from @ChrisAWalsh, Chen's and @Eunjung Alice Lee's labs:

Importantly, the mutation burdens and spectra that we estimate are overall consistent across data sets and single-cell sequencing technologies despite strong differences in seq depth, sequencing, etc:

Finally, we also show that the somatic mutations called by SComatic permit de novo discovery of mutational signatures! even in samples with relatively low mutational burdens (although in some cases the power is limited by low numbers of samples and mutations):

In sum, we present a novel algorithm that opens lots of possibilities to study somatic mutagenesis at single-cell resolution using healthy and diseased human samples, and also across the tree of life thanks to the advent of single-cell data for non-model organisms at eg @Chan Zuckerberg Biohub
We anticipate higher sensitivity to detect mutations as the throughput and seq depth of single-cell data sets increases. Novel platforms, such as MAS-Seq @PacBio, might increase sensitivity and specificity even further, opening avenues to map genotypes to transcriptomes at scale.
Thanks to everyone involved and to those who tirelessly generated (and annotated!!) large-scale single-cell data sets! As always, we cannot emphasise strongly enough the importance of good data sharing practices! SComatic is available at github.com/cortes-ciriano


Isidro Cortes-Ciriano
@isidrolauscher
Research Group Leader at EMBL-EBI @emblebi. @CR_UK Career Establishment Awardee. Cancer genomics.
Missing some tweets in this thread? Or failed to load images or videos? You can try to .